Neuroblastoma


Neuroblastoma (NB) is the commonest extracranial solid tumour of infancy. It is an embryonal tumour of the sympathetic nervous system. Tumours occur anywhere along the path of neural crest cell migration (adrenal medulla and sympathetic chain). The abdomen is the commonest site of occurrence (50% in the adrenal medulla and 25% para-spinal), followed by the thorax (20%), then the pelvis (~5%) and cervical region (~3%).
Around 100 new cases occur per year in the UK and it accounts for about 8% of all childhood tumours, but is responsible for almost twice that figure (15%) for tumour-related deaths – a truly malignant condition.
Symptoms
Symptoms can occur due to (i) its mass effect, (ii) metastatic disease, or from (iii) paraneoplastic syndromes. Most children are usually unwell at presentation. Abdominal tumours can cause pain and a palpable mass, para-spinal tumours can present with symptoms of spinal cord compression. Tumours in the chest can cause dyspnoea and chest pain or uncommonly present with pleural effusion. Lesions in the pelvis can present with constipation or urinary symptoms. Tumours involving the sympathetic ganglia in neck or thorax can cause Horner’s syndrome. Some children can present with proptosis, or racoon eyes, which can be an indication of skull-based disease.
About 40% of children will have metastatic disease at presentation with the usual sites being bone causing pain; bone marrow, causing anaemia and thrombocytopenia; lymph nodes; liver; and lungs.
Patients can develop a variety of paraneoplastic syndromes. These might include flushing, hypertension and tachycardia from catecholamine release; profuse watery diarrhoea from the release of vasoactive intestinal peptide; and opsoclonus-myoclonus syndrome which are spontaneous, rhythmic sideways movements of the eye, myoclonus and other cerebellar signs, and is thought to be due to anti-neuronal antibodies cross-reacting with the cerebellum.
Investigations
- Urine catecholamines – raised HMA/VMA
- Bloods: FBC/UE/CRP/LFT/LDH/ferritin
Imaging to assess the primary tumour & metastatic sites
- Ultrasound: A good first line modality to assess the primary tumour.
- Cross sectional imaging (CT/MRI): To assess the primary tumour and image defined risk factors (IDRF).
Image defined risk factors:
they have been used for staging and initial treatment decisions, and recently for anticipating surgical outcomes and predicting completeness of resection. They assess tumour extension into a second body compartment, encasement of any large blood vessels, trachea/large bronchial compression, involvement of major nerve roots, invasion of the spinal canal, or infiltration of the kidneys, mesentery, pericardium, liver, diaphragm, or pancreas. There are clear imaging guidelines on the assessment of IDRF – as these are very important to help determine the feasibility of surgery and document response to treatment. In recent studies, IDRFs at diagnosis were associated with larger, less differentiated, advanced stage, and higher risk NBL and at resection with increased operative difficulty and perioperative morbidity. However, the frequency of gross total resection and patient survival after resection were not associated with the presence of IDRFs .
- MIBG scan: Meta-iodobenzylguanidine. A radio-labelled molecule similar to noradrenaline (NA), is taken up by the NA transporter in 90% of neuroblastomas. This allows detection and monitoring of the primary tumour and metastases.
- CT chest to look for lung metastases
- SPECt-CT: involves combining CT and Isotope scanning.
- Bilateral bone marrow aspirates and trephine
Tissue diagnosis/Biopsy
Having tissue is extremely important in the staging and risk assessment for each patient. Treatment of neuroblastoma is tailored to the clinical and genetic characteristic of the individual tumour, therefore having a good sample of tissue for histopathology and cytogenetics is essential. This currently follows the published biopsy guidelines on the UKCCLG website (Children’s Cancer and Leukaemia Group, cclg.org.uk).
Tissue can be obtained in several ways
- Surgical biopsy (open or minimally invasive)
- Interventional Radiology – percutaneous needle core biopsy
Histology
Neuroblastoma is a stroma-poor tumour, containing mainly immature tissue (in comparison to ganglioneuroma, another neuroblastic tumour which comprises mainly mature ganglion cells, and is stroma-rich). Neuroblastoma can be undifferentiated, poorly differentiated or differentiating, depending on the degree of maturation of the tissue.
Tumour biology
The biology of the tumour has an increasingly important role in determining how it responds to treatment. By determining the different aspects of the tumour biology, we are now able to risk stratify the tumours and assign patients to specific treatment arms
- MYC-N amplification – this is one of the most important markers of disease severity
MYC-N is a proto-oncogene, located on the short arm of chromosome 2. MYC – N amplification (defined as having a more than a 4-fold increase in the number of MYC-N genes in relation to the number of copies of chromosome 2 is identified in 25% patients with NB, and in 40% of those with high-risk disease. Its presence is associated with a worse prognosis, is more aggressive and has a more rapid disease progression
- Numerical chromosomal anomalies: Diploid tumours have a poorer prognosis, compared to hyperdiploid tumours (gain of an odd number of chromosomes)
- Segmental chromosomal anomalies: 1p deletion, 17q deletion, 11q aberration. The presence of these confer a worse prognosis.
Staging
International Neuroblastoma Staging System (INSS)
Until about 2010, neuroblastoma was staged using the International Neuroblastoma Staging System (INSS). This was a post-surgical system, and depended on the degree of resection by the surgeon. The same tumour could be stage 1 or stage 3 depending on how extensively it was excised. It also did not allow for pre-operative planning, and made research and comparison between different centres difficult.
- Stage 1: localised tumour, completely resected
- Stage 2A: tumour completely resected, ipsilateral lymph nodes (LN) negative
- Stage 2B: tumour ± completely resected, ipsilateral LN positive
- Stage 3: Large tumour, incompletely resected, tumour crossing midline, contralateral LN positive
- Stage 4: metastatic disease
- Stage 4s: Children <12months, metastasis confined to liver, skin and BM (<10%)
International Neuroblastoma Research Group (INRG)
In 2009, the INRG introduced a new staging and risk stratification system. Tumours could be stratified pre-operatively, based on the presence or absence of specific image defined risk factors (IDRF).
INRG-SS (staging system)
- L1: well circumscribed tumour, localised to one body compartment. No IDRF
- L2: localised tumour with one or more IDRF
- M: metastatic disease
- Ms: neuroblastoma occurring in patients <18 months, tumour in skin, liver or bone marrow (<10%)
Risk stratification
 The INRG Task Force derived a risk stratification system for NB, based on the most significant and clinically relevant risk factors.
The INRG Task Force derived a risk stratification system for NB, based on the most significant and clinically relevant risk factors.
- INRG stage
- Age
- Histology
- Grade of tumour differentiation
- MYCN amplification
- 11q aberration
- Ploidy
 Poorly differentiated/undifferentiated histology, MYCN amplification, presence of 11q aberration and diploidy are all unfavourable biologic characteristics.
Poorly differentiated/undifferentiated histology, MYCN amplification, presence of 11q aberration and diploidy are all unfavourable biologic characteristics.
Based on the presence or absence of these characteristics the tumours can be stratified into low-, intermediate- and high-risk groups. These pre-treatment groups allow risk stratification, treatment planning and prognosis.
Event-free survival for:
- low-risk neuroblastoma is 75-85%,
- intermediate risk is 50-75%
- high risk neuroblastoma <50%.
Treatment for Low and intermediate risk disease
The patients are broadly divided into:
- Low Risk
- ≤ 18 months, L2 disease, non-MYCN amplified tumours
- ≤ 12 months with stage Ms
- Intermediate Risk
- ≤ 12 months with M (non-Ms) disease without MYCN amplification
- Children aged >18 months with L2 non-MYCN amplified tumours
These patients with low and intermediate risk tumours can be treated with observation alone – monitoring for tumour regression. If the tumour is L1, surgery alone may suffice, or surgery and differing lengths of chemotherapy based on the individual risk assessment. Treatment is guided by the CCLG guidelines, which are based on the SIOPEN LINES trial treatment recommendations.
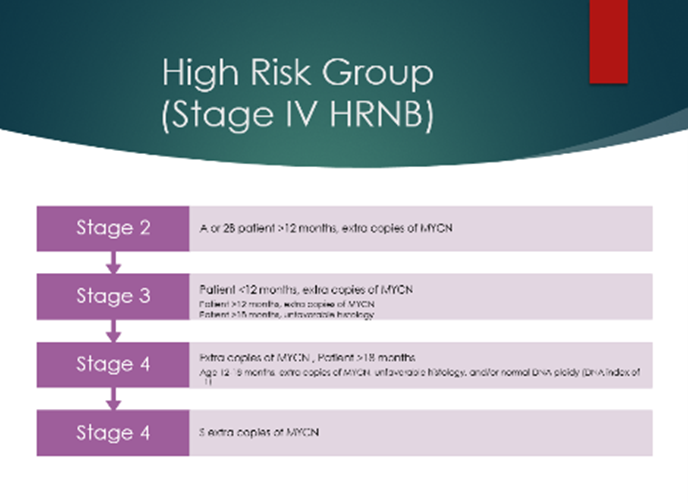
High risk neuroblastoma
This includes any patient with MYCN amplified neuroblastoma (other than stage L1) or any patient older than 12 months of age at diagnosis with stage M disease
Following confirmatory biopsy and tissue sampling, these patients are treated with a multimodal regime. Chemotherapy, stem cell rescue and transplant, surgery, radiotherapy and biological therapy. Despite the improvements in therapy, patients with high-risk disease still have a poor long-term outcome, with overall survival <50%.
Induction chemotherapy is used to decrease tumour burden, eliminate metastasis and facilitate safer surgical resection. Various induction regimens are used, in the UK we use Rapid COJEC. Following induction, peripheral blood stem cell harvest is performed followed by surgical excision of the tumour.
Surgical resection of the primary tumour is usually planned for the end of induction chemotherapy, as long as there is a good response. MRI and CT scan are required for planning and assessing operative IDRF. Recent evidence has shown that CT is superior to MRI for safe surgical planning as MRI can underestimate the extent of the disease compared to CT.
Surgical technique: Neuroblastoma is a heterogeneous tumour, and no single anatomic approach or technique can suffice for all. The tumour often encases major blood vessels, and a wide exposure that visualises and enables control of these is recommended. Careful, meticulous dissection is required. In extensive tumours a thoraco-abdominal approach has been described. Open surgery is the main stay for neuroblastoma, however minimally invasive surgery has been utilised in selected and localised tumours in the chest and abdomen with very good outcomes as recently shown by Gabra et al. (2021) in a multicentre SIOPEN study. Complete macroscopic excision, with an aim is to remove as much of the tumour as safely possible, without causing significant morbidity (which could delay further treatment) and mortality. There still remain differences in opinion as to how aggressively complete macroscopic excision should be sought.
.
Holmes et al (2020) recently published data from the HR-NBL1/SIOPEN study showing that complete excision (as assessed by the surgeon) was associated with a significantly higher 5-year event free and overall survival, compared to incomplete excision, and local recurrence was significantly lower. This was a retrospective study of 1,531 patients with HRNB. Complete excision was achieved in 77%, and occurred more often in patients with no IDRF and in those with adrenal primary disease. Operative mortality was 0.46%. There was a 9.7% rate of severe complications, which were significantly more likely to occur in those patients who achieved complete excision.
Von Allmen et al. (2017) showed that >90% resection had a benefit in local tumour control, however there was no improvement in outcome when patients had metastatic disease.
A systematic review and meta-analysis published in 2014 by Mullassery et al. showed that although there was a benefit to gross total resection in INSS stage 3 tumours, with improved 5 year survival, there was no difference in overall survival for stage 4 tumours undergoing gross total resection, and there were significant complications associated with this complex surgery.
Significant and serious complications occur in around 11 %, the operative mortality is around 0.7%
Following surgery patients will then undergo myeloablative therapy with autologous stem cell transplantation, and radiation to the primary tumour bed.
Maintenance therapy: 13-cis-retinoic acid/isotretinoin promotes tumour cell differentiation and slows the growth of neuroblastoma cells. Patients also receive immunotherapy with a monoclonal antibody called anti-GD2 antibody.
Treatment duration is approximately 18 months or longer if there are delays, and patients are followed up for 5 years following successful treatment.
Current/upcoming trials
- The SIOPEN High-Risk Neuroblastoma Clinical Trial 2 (HR-NBL2) is due to open early 2022.
- VERITAS study: Options for patients not achieving sufficient metastatic response after Rapid COJEC chemotherapy .
- BEACON neuroblastoma trial: To help establish the best chemotherapeutic approach for relapsed disease.
Further Reading
- Adkins ES, Sawin R, et al. Efficacy of complete resection for high-risk neuroblastoma: a Children’s Cancer Group study. J Pediatr Surg. 2004;39(6):931-6.
- Cohn SL, Pearson AD, et al. INRG Task Force. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009 Jan 10;27(2):289-97.
- Brisse HJ, McCarville MB, et al. International Neuroblastoma Risk Group Project. Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology. 2011 Oct;261(1):243-57.
- Mullassery D, Farrelly P, Losty PD. Does aggressive surgical resection improve survival in advanced stage 3 and 4 neuroblastoma? A systematic review and meta-analysis. Pediatr Hematol Oncol. 2014;31(8):703-16.
- von Allmen D, Davidoff AM, et al. Impact of extent of resection on local control and survival in patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol. 2017; 35:208-216.
- Phelps HM, Ndolo JM, et al. Association between image-defined risk factors and neuroblastoma outcomes. J Pediatr Surg. 2019;54: 1184-1191.
- Burnand K, Barone G, McHugh K, Cross K. Preoperative computed tomography scanning for abdominal neuroblastomas is superior to magnetic resonance imaging for safe surgical planning. Pediatr Blood Cancer. 2019 Nov;66(11):e27955.
- Holmes K, Pötschger U, et al. International Society of Paediatric Oncology Europe Neuroblastoma Group (SIOPEN). Influence of surgical excision on the survival of patients with stage 4 high-risk neuroblastoma: A Report From the HR-NBL1/SIOPEN Study. J Clin Oncol. 2020 ; 38(25):2902-2915.
- Gabra HO, Irtan S, et al. Minimally invasive surgery for neuroblastic tumours: A SIOPEN multicentre study: Proposal for guidelines. Eur J Surg Oncol. 2021: S0748-7983(21)00683-1.
Elizabeth O’Connor
Hany Gabra
[Edited by Mark Davenport – Oct 2021]